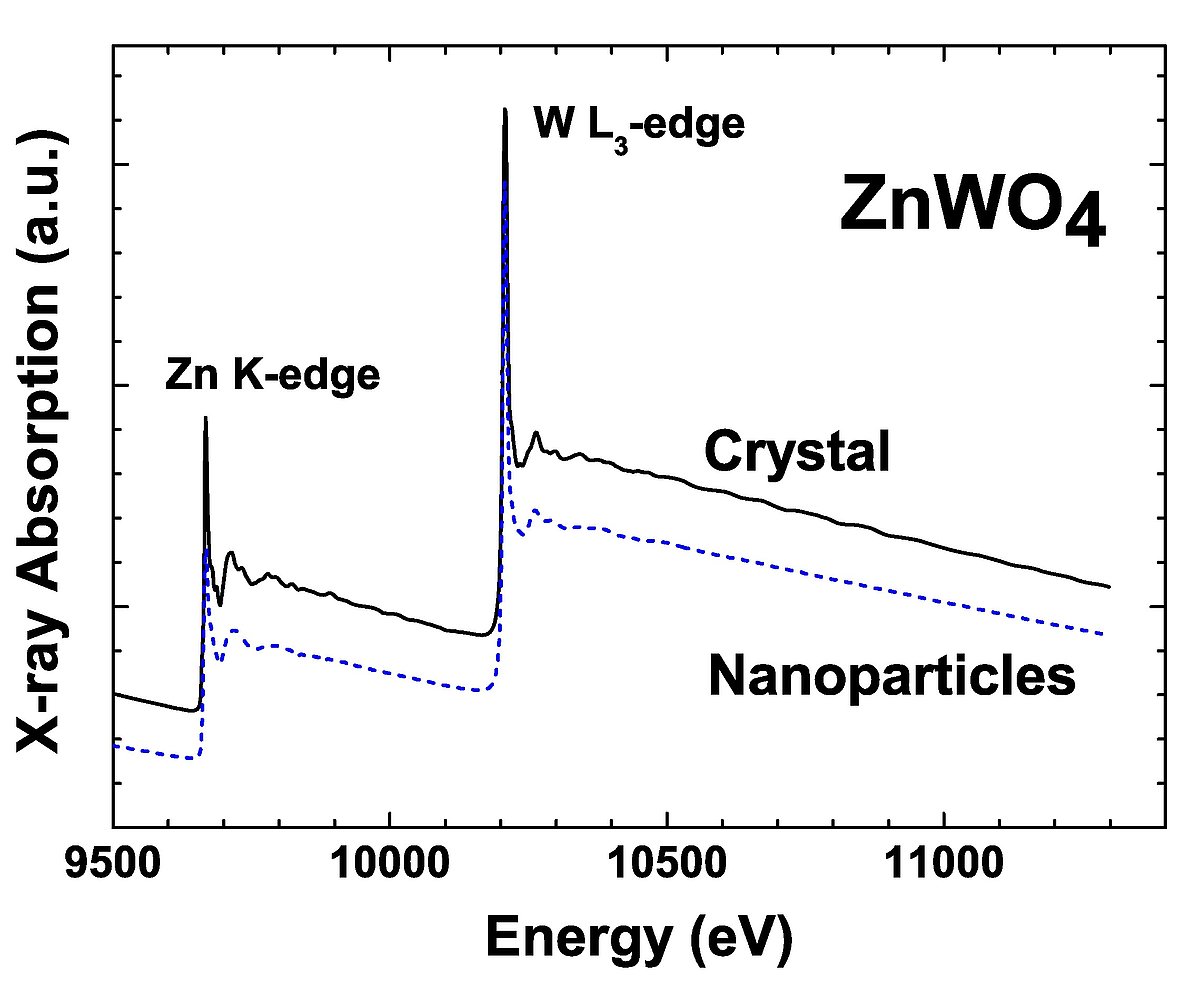
Rentgenabsorbcijas spektroskopija ir tieša lokālās struktūras pētīšanas metode. Tomēr mūsdienās rentgenabsorbcijas spektra izstieptās sīkstruktūras spektru analīzē pastāv grūtības daudzkārtīgās izkliedes efektu un termiskās nesakārtotības dēļ. Šajā darbā attīstīta jauna EXAFS spektru modelēšanas metode, kas balstās uz kvantu mehānikas un klasiskās molekulārās dinamikas (QMMD) pieeju, kas apvienota ar ab initio EXAFS spektru aprēķiniem. Metode ļauj iegūt unikālu informāciju par lokālo statisko un dinamisko atomāro struktūru kristāliskos materiālos, analizējot iegūtās daudzatomu sadalījuma funkcijas noteiktā temperatūrā un spiedienā. Šī metode tika lietota EXAFS spektru modelēšanai kristāliskiem ReO3, CaWO4 un ZnWO4 oksīdiem.
The x-ray absorption spectroscopy is widely used direct structure determination technique, which is highly sensitive to the local structure. However, the nowadays extended x-ray absorption fine structure (EXAFS) spectra analysis meets complications due to the presence of multiple scattering effects and thermal disorder. In this work we have developed the method of the EXAFS spectra analysis based on the quantum mechanics-classical molecular dynamics approach coupled with ab-initio EXAFS spectra calculations. The method allows one to obtain unique information on the local atomic static and dynamic structure in crystalline materials, through the analysis of the many-atom distribution functions at required temperature and pressure. The method was applied to the modeling of the EXAFS spectra from crystalline ReO3 , CaWO4 and ZnWO4. The investigation of nanocrystalline ZnWO4 is also reported.
{kind=link}